Table Of Content
Novel sequences and structures can be designed simultaneously by optimizing the loss function through Monte Carlo–simulated annealing. Diverse structures were designed by the model and shown to be folded by experimental characterization. De novo protein design seeks to generate proteins with specified structural and/or functional properties, for example, making a binding interaction with a given target12, folding into a particular topology13 or containing a catalytic site4. Denoising diffusion probabilistic models (DDPMs), a powerful class of machine learning models recently demonstrated to generate new photorealistic images in response to text prompts14,15, have several properties well suited to protein design. First, DDPMs generate highly diverse outputs, as they are trained to denoise data (for instance, images or text) that have been corrupted with Gaussian noise.
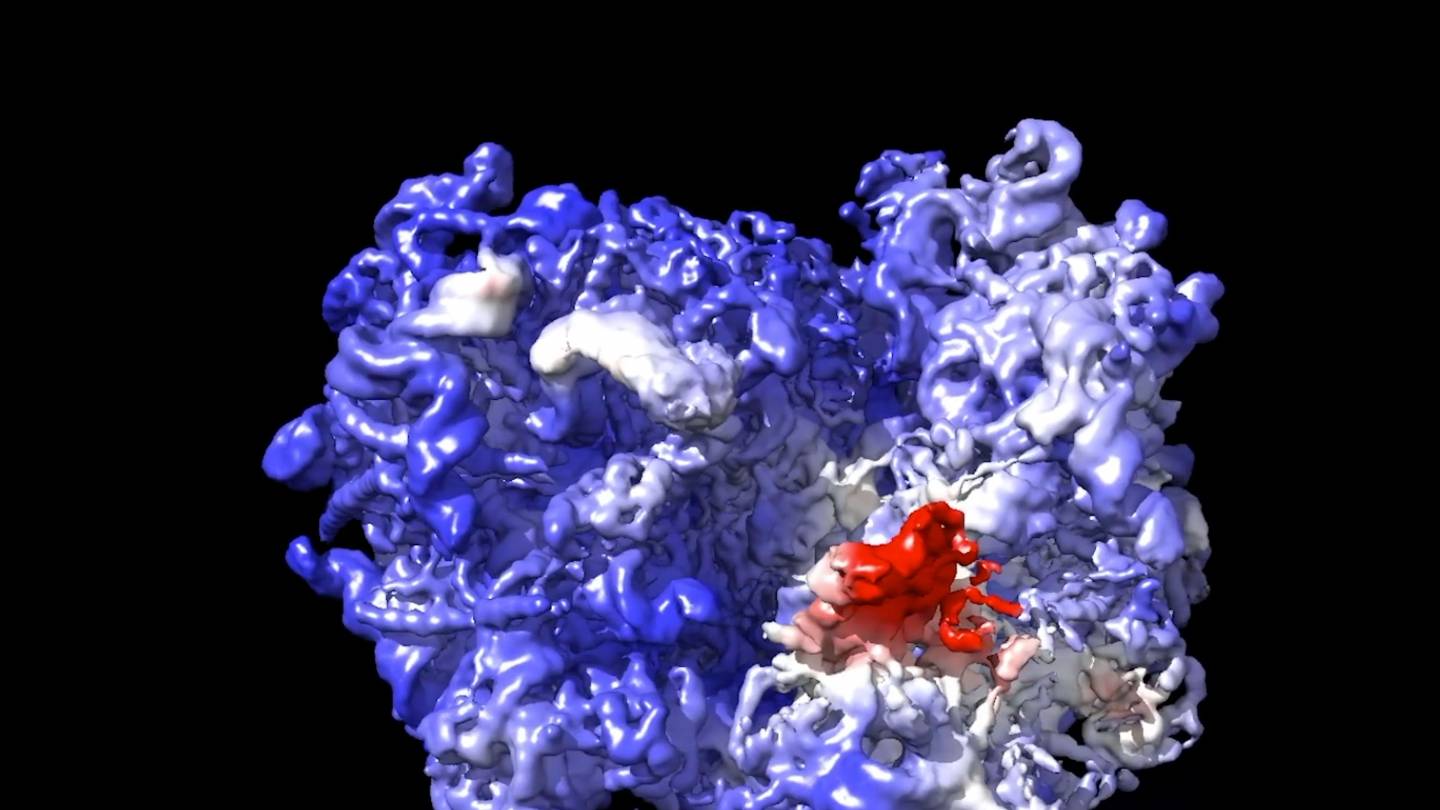
Extended Data Fig. 9 Cryo-electron microscopy structure determination of designed Influenza HA binder.
Immunoglobulins with defined complementarity determining regions can then be generated through latent space sampling. A new method used the idea of neural network “hallucination” (generation of structures) for the protein design (45). The TR-Rosetta network is a fast method to predict the inter-residue contact map of an arbitrary sequence. A loss function is defined as Kullback-Leibler divergence (84) between the TR-Rosetta neural network–predicted contact map and a background distribution.
Optimization algorithms without guarantees
H.E.E., A.J.B., R.J.R., L.F.M., B.I.M.W., S.J.P., N.H., A.C., S.V.T., J.L.W. and B.L.T. experimentally characterized designs. Agree that the order of their respective names may be changed for personal pursuits to best suit their own interests. To fully understand protein function in the cellular milieu, it is desirable to be able to study and manipulate protein activity in living cells. Since Green Fluorescent Protein (GFP) was first cloned two decades ago,59 fluorescent proteins have become a powerful and omnipresent tool in biology. The potential of GFP to study protein expression and localization was recognized early on, but several important limitations had to be overcome before it could be used routinely in biological applications. Protein design has played an important role in overcoming these obstacles and in extending the in vivo potential of fluorescent proteins.
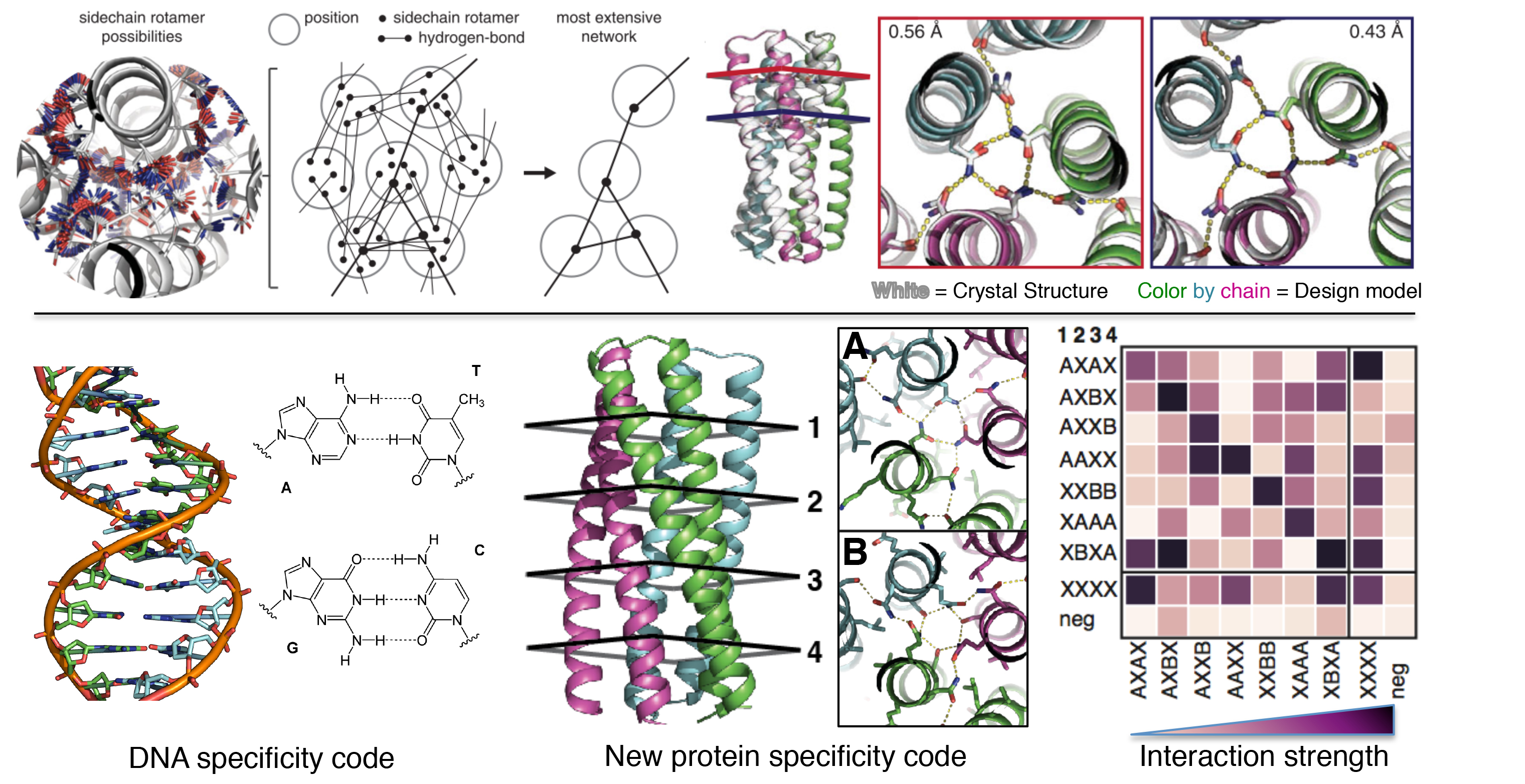
Hydrogen-bonding networks
ProRefiner: an entropy-based refining strategy for inverse protein folding with global graph attention - Nature.com
ProRefiner: an entropy-based refining strategy for inverse protein folding with global graph attention.
Posted: Thu, 16 Nov 2023 08:00:00 GMT [source]
Physics-based energy functions typically model an attractive-repulsive Lennard-Jones term between atoms and a pairwise electrostatics coulombic term[17] between non-bonded atoms. To create the algorithm, the scientists trained an AI model with information from hundreds of thousands of known interactions between chemical molecules and the corresponding three-dimensional protein structures. Now, without human intervention, a generative AI is able to develop drug molecules from scratch that match a protein structure. This groundbreaking new process ensures right from the start that the molecules can be chemically synthesised. In addition, the algorithm suggests only molecules that interact with the specified protein at the desired location and hardly at all with any other proteins.
Finally, the structural modeling verification experiment indicates that the sequences designed by SPDesign can fold into the native structures more accurately. Machine learning models trained with the rich structural data from the PDB are able to generate novel protein backbone structures (Fig. 2E). A generative adversarial network (81) model builds protein structures represented as pairwise distances between all backbone atoms.
Specificity of ligand binding is often realized by polar interactions which are highly sensitive to the positions and orientations of polar groups. A misaligned hydrogen bond could cause a considerable free energy penalty and reduce the binding affinity by an order of magnitude. Early studies designed de novo binding sites by manually defining side chains that form favorable interactions with ligands (11, 20, 26). An effort that uses HBNet and a Monte Carlo sequence design algorithm to design hydrogen bonds resulted in designs that bind to ligands, but a crystal structure revealed that the ligand is rotated 180° in the pocket around a pseudo-two-fold axis in the compound (137).
By studying the genetic code and the mechanisms underlying protein synthesis, researchers can manipulate the sequences of amino acids that make up a protein. This knowledge enables scientists to create proteins with specific functions, such as binding to target molecules, catalyzing chemical reactions, or forming stable structures. Toll-like Receptor 3 (TLR3) is a pattern recognition receptor that initiates antiviral immune responses upon binding double-stranded RNA (dsRNA). Several nucleic acid-based TLR3 agonists have been explored clinically as vaccine adjuvants in cancer and infectious disease, but present substantial manufacturing and formulation challenges.
De novo design of protein structure and function with RFdiffusion - Nature.com
De novo design of protein structure and function with RFdiffusion.
Posted: Tue, 11 Jul 2023 07:00:00 GMT [source]
Computational Redesign of Metalloenzymes for Catalyzing New Reactions
The Foldit developers experimentally tested 146 top designs and identified 56 designs that adopted well-folded monomeric structures. The experimentally solved structures of four of these designs closely agreed with the computational models. A workaround to the difficulty of de novo backbone design is redesigning native backbone structures from the PDB for new functions (18, 19, 20). Because proteins are not static, state-of-the-art design methods typically consider small structural adjustments in response to sequence changes, or to diversify native backbones. In particular, several approaches have been developed to mimic “back-rub” motions (49, 50), a common mechanism for interconverting between alternate backbone conformations observed in high-resolution (≤1 Å) crystal structures (51).
Protein design articles from across Nature Portfolio
To address these problems, state-of-the-art side-chain design methods sample both side-chain rotamers and local backbone conformations (50, 52, 94, 95) (Fig. 3C). Typically, methods that exploit backbone flexibility or use backbone ensembles outperform the fixed backbone design (96, 97). A study benchmarked (98) several flexible backbone side-chain design methods including CoupledMoves (94), BackrubEnsemble (56), and FastDesign compared with a fixed backbone design method using the same scoring function. Methods that simultaneously, rather than sequentially, optimize sequence and backbone structure, such as CoupledMoves (94), may be advantageous (98). Backbone structures determine the overall shapes of proteins and therefore play a critical role in protein functions.
The advances not only highlight design goals reachable now but also point to the challenges and opportunities for the future of the field. RFdiffusion readily generates diverse unconditional designs up to 600 residues in length that are accurately predicted by AF2, far exceeding the complexity and accuracy achieved by most previous methods (a recent Hallucination-based approach also achieved high unconditional performance53). Half of our tested unconditional designs express in a soluble way, and have circular dichroism spectra consistent with the design models and high thermostability. Despite their substantially increased complexity, the ideality and stability of RFdiffusion designs is akin to that of de novo protein designs generated using previous methods such as Rosetta. RFdiffusion enables generation of higher-order architectures with any desired symmetry, unlike Hallucination methods, which have so far been limited to cyclic symmetries.
Combining machine learning models and traditional Monte Carlo samplers improves performance over every single method (103, 109). 3c,d, starting from random noise, RFdiffusion can readily generate elaborate protein structures with little overall structural similarity to structures seen during training, indicating considerable generalization beyond the PDB (see Supplementary Table 1 for a comparison of all designs in the paper to the PDB). The designs are diverse (Supplementary Fig. 3a), spanning a wide range of alpha, beta and mixed alpha–beta topologies, with AF2 and ESMFold (Fig. 2c, Extended Data Fig. 1b,c and Supplementary Fig. 2b) predictions very close to the design structure models for de novo designs with as many as 600 residues. RFdiffusion generates plausible structures for even very large proteins, but these are difficult to validate in silico as they are probably generally beyond the single sequence prediction capabilities of AF2 and ESMFold. The quality and diversity of designs that are sampled are inherent to the model, and do not depend on any auxiliary conditioning input (for example, secondary structure information8).
The rotamer interaction field (RIF) docking method (29) generates an ensemble of billions of discrete amino acid side chains that make hydrogen-bonding and hydrophobic interactions with the target ligand. The method then searches for protein backbone scaffolds that are able to present ligand-binding side chains with the appropriate geometry. RIF docking was successfully applied to design a binding site for the fluorogenic compound DFHBI into a de novo beta barrel scaffold (29). Two other methods use the structural information in the PDB to generate binding-site ensembles (25, 138). These methods break the ligand into smaller substructures (fragments) and find protein residues that interact with the ligand fragments from the PDB.
Protein design and engineering are extremely difficult topics that require a high level of skill to achieve the intended outcomes. Protein engineering procedures require accurate result interpretation, and researchers also need to be able to evaluate experimental findings and compare them to the hypothesis of the experiment. Furthermore, the proteins generated via engineering have particular uses, including vaccine development, gene therapy, medication administration, antibody engineering, and enzyme modification. The complex sequences of DNA and amino acids must be changed in order to create proteins that perform the desired function.

No comments:
Post a Comment